
 Definition
Definition
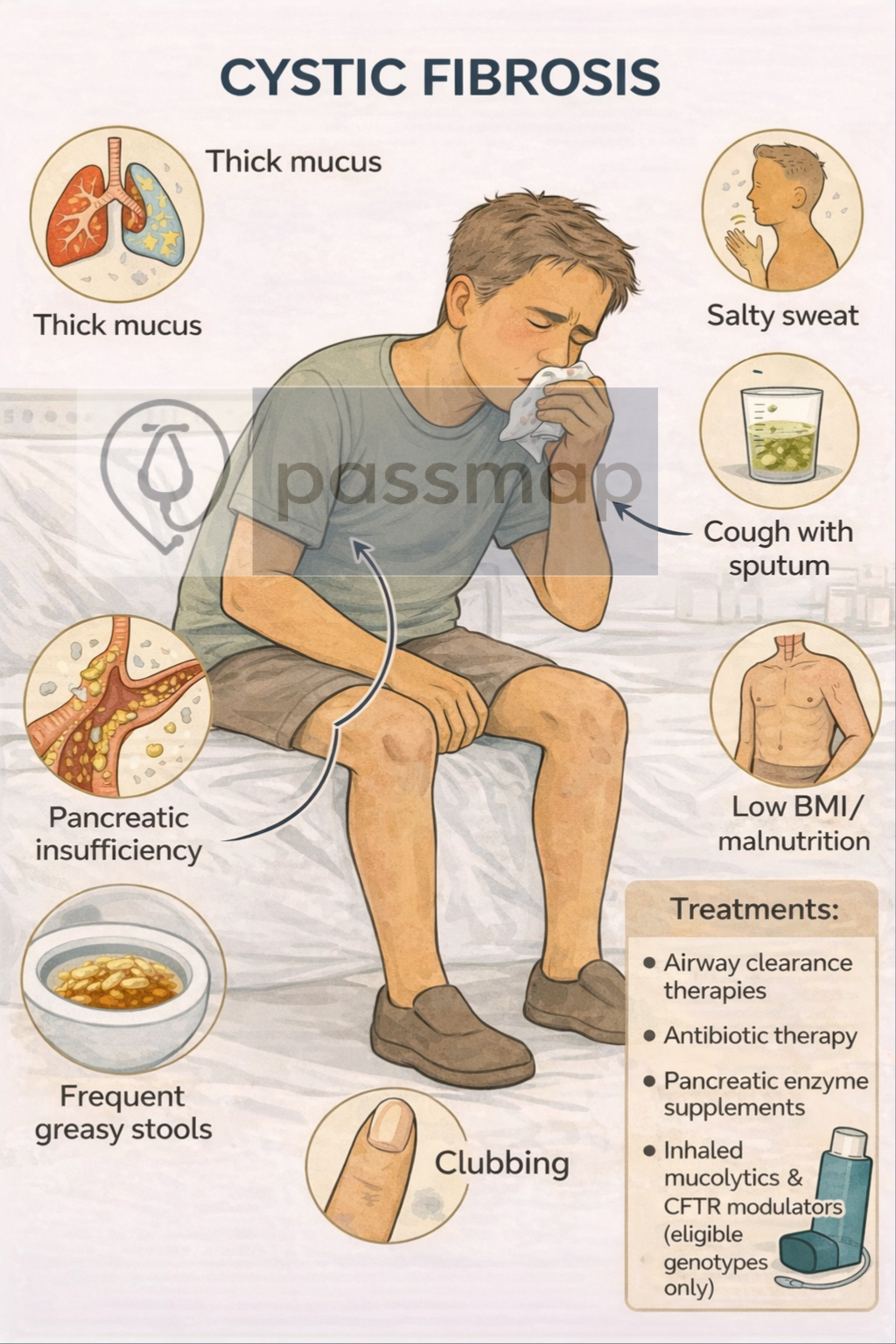
Cystic Fibrosis is an autosomal recessive genetic disorder caused by mutations in the CFTR gene, leading to defective chloride transport and production of thick, sticky secretions affecting the lungs, pancreas, gastrointestinal tract, and reproductive system.
 EXAM ANCHOR – CORE CONCEPT (PARA)
EXAM ANCHOR – CORE CONCEPT (PARA)
- CF = autosomal recessive CFTR mutation
- Defective chloride transport → thick, sticky secretions
- Multisystem disease (lungs, pancreas, GI, reproductive)
 PARA commonly asks:
PARA commonly asks:
Which genetic disorder causes thick, sticky secretions due to CFTR dysfunction?
🎯 EXAM ANCHOR – INHERITANCE
Autosomal recessive inheritance
Both parents must be carriers
Siblings at increased risk
PARA commonly asks:
What is the mode of inheritance of cystic fibrosis?
 Physical Examination Findings
Physical Examination Findings
- Chronic productive cough
- Coarse crackles ± wheeze
- Finger clubbing
Signs of advanced disease:
Pulmonary hypertension
Cor pulmonale
EXAM ANCHOR – IMAGING (PARA)
Sweat chloride test = diagnostic gold standard
Elevated sweat chloride confirms CF
Genetic testing supports diagnosis
PARA commonly asks:
Answer: Bronchiectasis with mucus plugging
EXAM ANCHOR – MICROBIOLOGY
PARA commonly asks:
- Commonest organism in children: Staphylococcus aureus (often managed with prophylactic Flucloxacillin). Commonest in adults: Pseudomonas aeruginosa.”
Which organism in adults is most commonly colonises the lungs in cystic fibrosis?
 Management
Management
 Mnemonic: CLEAN AIRWAYS
Mnemonic: CLEAN AIRWAYS
Chest physiotherapy
Daily airway clearance is essential
Cornerstone of long-term management
EXAM ANCHOR – PANCREATIC INVOLVEMENT
Pancreatic insufficiency is common
Causes steatorrhoea & weight loss
Requires enzyme replacement
PARA commonly asks:
What causes steatorrhoea and weight loss in cystic fibrosis?
 Key PARA Exam Traps – Cystic Fibrosis (CF)
Key PARA Exam Traps – Cystic Fibrosis (CF)
 Autosomal recessive CFTR mutation → thick, sticky secretions
Autosomal recessive CFTR mutation → thick, sticky secretions
Gold standard test = sweat chloride (↑ chloride)
Most common respiratory pathogen = Pseudomonas aeruginosa
Chronic lung disease → bronchiectasis (not reversible)
Pancreatic insufficiency → steatorrhoea → treat with Creon
Male infertility common (congenital absence of vas deferens)
Meconium ileus in neonates = CF until proven otherwise
CF-related diabetes is common
Daily airway clearance is core management, not optional
CFTR modulators only work for eligible genotypes
 The “Must-Know” PARA Numbers for CF
The “Must-Know” PARA Numbers for CF
| Factor | PARA / NICE Standard |
| Sweat Test | Chloride > 60 mmol/L |
| Diabetes Screen | OGTT (not HbA1c) annually from age 10 |
| Inheritance | Autosomal Recessive (25% chance if both parents are carriers) |
| Microbiology | S. aureus (Child) → P. aeruginosa (Adult) |
| The “Social” Rule | Patients with CF must never meet in person (risk of cross-infection with Burkholderia cepacia). |
Last updated in line with NICE NG78 (2017), reviewed 2023
Last reviewed: February 2026 PASSMAP ensures all content is NICE-aligned and reviewed for Physician Associate Registration Assessment (PARA) success.
PASSMAP ensures all content is NICE-aligned and reviewed for Physician Associate Registration Assessment (PARA) success.
Educational platform. Not medical advice.